 9559179325
9559179325 9559179325
9559179325 Login
Login
 9559179325
9559179325


Overview of FDA Certification
The Food and Drug Administration (FDA), which was founded in 1906 after the enactment of the Federal Food and Drugs Act, is a government agency. It is the oldest consumer protection agency in the world. The FDA certification is required before the products can be sold in the United States.
It is a division of the Department of Health and Human Services in the United States. The FDA's primary role is to safeguard public health and associated authorities by ensuring the safety and security of human and biologically derived products. Biological products, medical services, cosmetics, prescription and non-prescription medications, veterinary medications, tobacco, and other radiation emitting products are all regulated by the FDA.
Following an inspection by an FDA official, the FDA issues Form 483 for FDA Certification of the items. According to FDA rules, food and pharmaceuticals manufacturing facilities in India must follow Current Good Manufacturing Practices (CGMP). FDA assists in ensuring the safety, quality, and efficacy of medical and food goods manufactured in India for export to the United States.
What is FDA certification and how does it work?
The Food and Drug Administration (FDA) is a federal agency under the Department of Health and Human Services in the United States. The FDA's primary role is to safeguard public health and associated authorities by ensuring the safety and security of human and biologically derived products.
Biological products, medical services, cosmetics, prescription and non-prescription medications, veterinary medications, tobacco, and other radiation emitting products are all regulated by the FDA.
CDSCO and FDA have a lot in common.
The Central Drugs Standard Control Organization (CDSCO), which is equivalent to the FDA in the United States and is under the Directorate General of Health Services, Ministry of Health & Family Welfare, and Government of India, is India's national regulatory authority for pharmaceuticals and medical devices.
The Drug Controller General of India (DCGI) is one of the CDSCO departments in charge of overseeing and regulating the manufacture, authorization, and sale of medical services, as well as medications and equipment, in India.
Clinical research, professional examination of the effect on human health, and enforcement under the Drugs and Cosmetics Act are all examples of this. Inspection, audit, and surveillance are carried out by the DCGI department through various zonal units across India.
What Kinds of Products Do You Need to Get FDA Approval?
FDA approval is conditional on the sort of product being sold in the United States. FDA certification is not required for all types of items. The goods that require FDA certification are listed below.
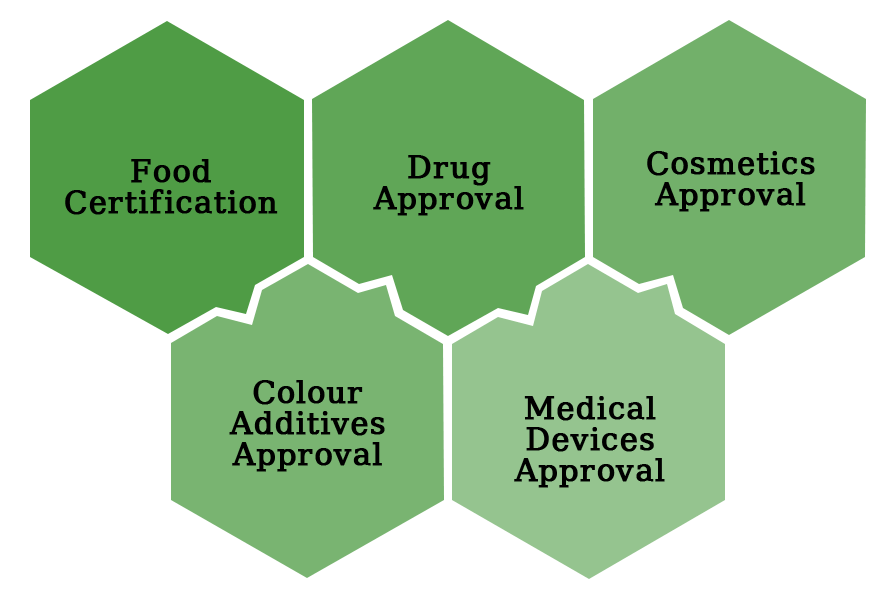
Food Certification by the FDA
Food products do not need FDA certification, but food processing facilities must register with the FDA. The FDA does not require any form of certification for the product before it may be distributed in the United States.
Officials from the Food and Drug Administration (FDA) travel to India to inspect food and pharmaceutical manufacturing facilities. The official verifies that the plants are in accordance with the FDA's guidelines.
Drug Approval from the FDA
The FDA examines the drug to see if it complies with the Over-the-Counter (OTC) monograph. Drugs must be both safe and effective. The OTC monograph specifies the parameters under which the drug items are safe and effective to use. monograph (OTC)
On the other hand, if the new medicine does not conform with the OTC monograph, FDA certification will be required. Drug makers undergo lab, human, and animal testing before submitting the results to the FDA. The FDA will analyze the information given and may grant the product FDA certification.
Cosmetics with FDA Approval
Cosmetics and its ingredients (apart from Color Additives) do not require FDA approval before being marketed. Cosmetics companies are not required to register with the FDA, but their products must be safe for customers to use.
Certain cosmetic claims may cause the FDA to regulate the cosmetic product as a drug product, and in some situations, this may result in the cosmetic product receiving an FDA approval certificate.
Color Additives Have Been Approved by the FDA
FDA certification is required for colour additives used in food, drugs, some medical devices, and cosmetics. Color batch certification from the FDA is required for some high-risk colours. The use of colour additives is limited to FDA-approved compliances. The FDA has placed restrictions on dangerous colour additives.
Medical Devices That Have Been Approved By The FDA
The FDA categorizes medical devices into three risk categories. The following are the categories: Class I, Class II, and Class III are the three levels of classification. Medical gadgets in the Class III category are the most dangerous. Only FDA certification is required for these Class III devices. Class III medical device makers should reassure the FDA that their products are safe and effective to use.
Class I
Medical devices have a low chance of harming the user. Elastic bandages, face masks, and tongue depressors are among the devices available. Class I medical devices account for 47% of all medical devices, and the vast majority of them (95%) are exempt from regulatory oversight. This means that most items in this category are exempt from the FDA's premarket approval process.
Class II
Medical devices are considered to be a medium-risk item. This group covers anything from motorized wheelchairs to some pregnancy test kits to the Apple Watch ECG app, and it accounts for 43% of all devices. Although some Class II devices are exempt from regulatory requirements, the majority must submit a 510(k), or premarket notification, at the very least.
Class III
Medical equipment are the ones that put a patient's life in jeopardy the most. This group includes only 10% of all gadgets. These are devices that are implanted in the body, are life-sustaining, or pose an undue danger in any other way. Pacemakers, breast implants, and replacement heart valves are examples of Class III devices. These devices must submit a Premarket Approval application (PMA).
Medical device makers should register with FDA and supply FDA with a full list of their gadgets. The FDA does not certify firms or devices based on their registration and listing with FDS.
What Are the Advantages of FDA Approval?
The advantages of FDA Approval are as follows-

The Effect on the Industry
FDA requirements are stringent, and obtaining FDA approval can take a long time, a series of iterations, and testing. However, once you have it, this product will have a significant impact in the direction of the consumer, as it will be available for purchase, thus improving the lives, conditions, and health of your target market.
In-Demand Product
In terms of demand, products that have received FDA approval quickly rise to the top. Consumers realise that it has been well tested and is safe to use. As a result, demand for your product will increase. Having the FDA's seal of approval is more often than not a necessity rather than a pleasant to have.
Gaining Access to Global Markets
When the FDA approves your product, you'll also receive a Certificate of Foreign Government Approval (CFG). This effectively allows you to expand your deal points and begin selling to countries such as Japan, Brazil, Australia, and China. This shows other nations where your product has been approved by the FDA and can be marketed and exported from the United States.
Boost your credibility
FDA is well-known over the world, and it has developed its own brand. The FDA has a steadfast concept and a comprehensive number of strategies to assist a company in obtaining FDA approval. If your product receives FDA approval, it has undergone extensive product quality testing and different enhancement techniques before being submitted to the FDA for review.
Extend the scope of your company's expertise
It will be much easier for your organization to expand into different areas of business after you obtain FDA a. After your product has been approved by the FDA, the next step is to constantly update, upgrade, and improve it, which may include branching out into other industries.
Improved Concept to Help the Product Advance
It's about having a better understanding of the steps involved in creating, designing, testing, and manufacturing a high-quality product. Fundamentally, it's all about demonstrating safety and efficacy problems at every turn.
Establishes Funding Lines
The advantage of FDA approval is that you will have better access to finance. The product manufacturer's FDA clearance shows that they are a strict and approved company. Big companies are constantly on the search for smaller businesses in need of finance to add to their product line.
Recognized by Medical Networks
Medical facilities and hospitals will only consider a food or medical product if it has received FDA approval. You can't even sell your products in pharmacies without it. You'll very certainly be working with medical facilities on clinical trials, pilot testing, or usability devices as you develop your product. This is a fantastic method to grow and broaden your network.
What are the Documents Required for FDA Certification?
The documents required are as follows-
Entry Documentation
- Airway Bill
- Bill of Lading
- Invoice
- Purchase Order
Commodity Specific Documentation
- Any other document as requested.
- Documentation stating the identity of the actual owner of the product
- Growers List
- Labelling Copies
- Packing List
- The statement of the intended use or end use of the product
What is the procedure for getting FDA approval?
The FDA has the ability to investigate other countries' facilities that supply food and drug items to the US. The FDA's Current Good Manufacturing Practices shall be followed by all food and medicine manufacturing facilities in India that export to the United States (CGMP). The FDA certification procedure is as follows: -
- Food and medicine plants in other nations are visited by FDA officials.
- Following the completion of the inspection, an FDA official issues Form 483 to the manufacturers.
- If officials discover any deviations from FDA requirements, they should report them on Form 483.
- The areas where there is a deviation from regulatory expectations shall be specified on Form 430.
- The modifications are then presented and reviewed with the manufacturing company's management.
- An EIR is also issued by the FDA (Establishment Inspection Report). The EIR determines whether or not a particular action should be taken. In the following order, the technique is carried out: -
- Pre-clinical evaluation.
- Look into a new application.
- FDA Approval
- Within 15 working days, FDA should receive your response to Form 438. The response should be thorough, including justifications for the flaws. The response should also include a fresh action plan to address the weaknesses.
- Following the submission of the Form 438 response, FDA officials may send a warning letter. If FDA officials are not convinced by the Form 438 answer, they will send a warning letter.
- The warning letter response should be satisfactory and sent within 15 working days. If the response is poor, the manufacturer's licence may be revoked, product approval may be withheld, and products may be placed on import alert.
- The FDA grants approval for drug importation if the FDA officials are satisfied.
If FDA authorities discover that some observations made while inspecting a food and drug plant are of a serious character, they may send a warning letter without first issuing Form 438.
What are the Additional Regulatory Organizations?

CDHR - Center For Devices and Radiological Health
The Center For Devices and Radiological Health (CDRH) is a research and development organisation that focuses on medical devices and radiological health. The Center for Devices and Radiological Health (CDRH) is a regulatory agency within the Food and Drug Administration (FDA) of the United States Department of Health and Human Services. They are responsible for enforcing and implementing the laws and regulations that apply to radiation-producing electronic equipment and medical devices that include lasers and light devices.
IEC- International Electro Technical Commission
The International Electro Technical Commission (IEC) is a body that regulates the electrical industry. The IEC (International Electrotechnical Commission) is an international standards body that develops and publishes international standards for all electrical, electronic, and related technologies, which are commonly referred to as "electrotechnology."
Why Choose Us

Free Legal Advice

Transparent Pricing

On Time Delivery

Expert Team

Money Back Guarantee

200+ CA/CS Assisted

Lowest Fees

Easy EMIs
Frequently Asked Questions
When FDA officials observe some conditions that are in violation of FDA rules at the conclusion of an inspection, they issue Form 483. All observations in Form 483 must be mentioned clearly, specifically, and significantly by FDA authorities.
The FDA's Form 483 informs the company's management of the undesirable conditions. Form 438 is presented and reviewed with the company's senior management when the inspection is completed. Form 438 should be responded to in writing by the company's management. The correct action plan should be included in the form.
Following the examination, Form 438 is discussed with the company's management. The FDA authorities' observations in Form 438 are read and discussed in order to gain a thorough grasp of what they are and what they represent.
The FDA can receive the entrance documentation in one of the following ways:
ITACS can be used to upload the papers (Import Trade Auxiliary Communication System). The customs broker, importer, or any other responsible party can upload the paperwork; or
Locate the import office's contact page and seek for the local import division's postal address, email address, and/or fax number for document submission.












